RNA Enzymes
Molecular Dynamics Study of Twister Ribozyme: Role of Mg2+ Ions and the Hydrogen-Bonding Network in the Active Site
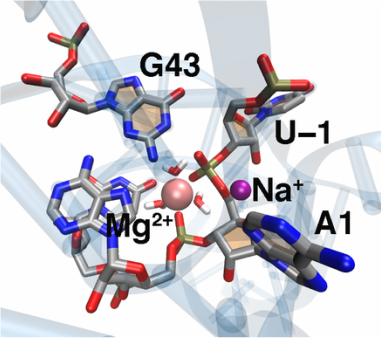
The recently discovered twister ribozyme is thought to utilize general acid–base catalysis in its self-cleavage mechanism, but the roles of nucleobases and metal ions in the mechanism are unclear. Herein, molecular dynamics simulations of the env22 twister ribozyme are performed to elucidate the structural and equilibrium dynamical properties, as well as to examine the role of Mg2+ ions and possible candidates for the general base and acid in the self-cleavage mechanism. The active site region and the ends of the pseudoknots were found to be less mobile than other regions of the ribozyme, most likely providing structural stability and possibly facilitating catalysis. A purported catalytic Mg2+ ion and the closest neighboring Mg2+ ion remained chelated and relatively immobile throughout the microsecond trajectories, although removal of these Mg2+ ions did not lead to any significant changes in the structure or equilibrium motions of the ribozyme on the microsecond time scale. In addition, a third metal ion, a Na+ ion remained close to A1(O5′), the leaving group atom, during the majority of the microsecond trajectories, suggesting that it might stabilize the negative charge on A1(O5′) during self-cleavage. The locations of these cations and their interactions with key nucleotides in the active site suggest that they may be catalytically relevant. The P1 stem is partially melted at its top and bottom in the crystal structure and further unwinds in the trajectories. The simulations also revealed an interconnected network comprised of hydrogen-bonding and π-stacking interactions that create a relatively rigid network around the self-cleavage site. The nucleotides involved in this network are among the highly conserved nucleotides in twister ribozymes, suggesting that this interaction network may be important to structure and function.
The recently discovered twister ribozyme is thought to utilize general acid–base catalysis in its self-cleavage mechanism, but the roles of nucleobases and metal ions in the mechanism are unclear. Herein, molecular dynamics simulations of the env22 twister ribozyme are performed to elucidate the structural and equilibrium dynamical properties, as well as to examine the role of Mg2+ ions and possible candidates for the general base and acid in the self-cleavage mechanism. The active site region and the ends of the pseudoknots were found to be less mobile than other regions of the ribozyme, most likely providing structural stability and possibly facilitating catalysis. A purported catalytic Mg2+ ion and the closest neighboring Mg2+ ion remained chelated and relatively immobile throughout the microsecond trajectories, although removal of these Mg2+ ions did not lead to any significant changes in the structure or equilibrium motions of the ribozyme on the microsecond time scale. In addition, a third metal ion, a Na+ ion remained close to A1(O5′), the leaving group atom, during the majority of the microsecond trajectories, suggesting that it might stabilize the negative charge on A1(O5′) during self-cleavage. The locations of these cations and their interactions with key nucleotides in the active site suggest that they may be catalytically relevant. The P1 stem is partially melted at its top and bottom in the crystal structure and further unwinds in the trajectories. The simulations also revealed an interconnected network comprised of hydrogen-bonding and π-stacking interactions that create a relatively rigid network around the self-cleavage site. The nucleotides involved in this network are among the highly conserved nucleotides in twister ribozymes, suggesting that this interaction network may be important to structure and function.
DNA Repair via Translesion Synthesis
Relative Binding Affinities of Adenine and Guanine to Damaged and Undamaged DNA in Human DNA Polymerase η: Clues for Fidelity and Overall Efficiency
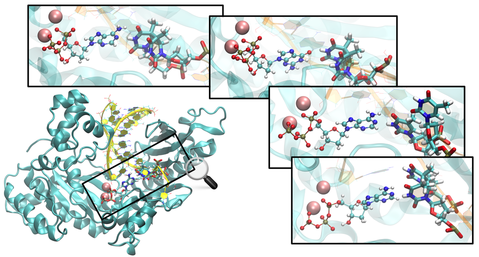
Human DNA polymerase η (Pol η) plays an essential protective role against skin cancer caused by cyclobutane thymine-thymine dimers (TTDs), a frequent form of DNA damage arising from exposure to the sun. This enzyme rescues stalled replication forks at the TTDs by inserting bases opposite these DNA defects. Herein we calculate binding free energies for a free deoxyribose nucleotide triphosphate, dATP or dGTP, to Pol η complexed with undamaged or damaged DNA. The calculations indicate that the binding of dATP to the enzyme-DNA complex is thermodynamically favored for TTD-containing DNA over undamaged DNA, most likely because of more extensive hydrogen-bonding interactions between the TTD and the enzyme that hold the TTD more rigidly in place. The calculations also illustrate that dATP binding is thermodynamically favored over dGTP binding at both thymine positions of the TTD, most likely due to more persistent and stable hydrogen-bonding interactions between the TTD and dATP than between the TTD and dGTP. This free energy difference is slightly greater for binding at the 5’ thymine position than at the 3’ thymine position, presumably because of stabilization arising from the A:T base pair formed at the 3’ position of the TTD in the previous step of Pol η function. All of these trends in binding free energies are consistent with experimental measurements of binding strength, fidelity, processivity, and overall efficiency. The insights gained from this analysis have implications for drug design efforts aimed at modifying the binding properties of this enzyme for improving cancer chemotherapy treatments.
Human DNA polymerase η (Pol η) plays an essential protective role against skin cancer caused by cyclobutane thymine-thymine dimers (TTDs), a frequent form of DNA damage arising from exposure to the sun. This enzyme rescues stalled replication forks at the TTDs by inserting bases opposite these DNA defects. Herein we calculate binding free energies for a free deoxyribose nucleotide triphosphate, dATP or dGTP, to Pol η complexed with undamaged or damaged DNA. The calculations indicate that the binding of dATP to the enzyme-DNA complex is thermodynamically favored for TTD-containing DNA over undamaged DNA, most likely because of more extensive hydrogen-bonding interactions between the TTD and the enzyme that hold the TTD more rigidly in place. The calculations also illustrate that dATP binding is thermodynamically favored over dGTP binding at both thymine positions of the TTD, most likely due to more persistent and stable hydrogen-bonding interactions between the TTD and dATP than between the TTD and dGTP. This free energy difference is slightly greater for binding at the 5’ thymine position than at the 3’ thymine position, presumably because of stabilization arising from the A:T base pair formed at the 3’ position of the TTD in the previous step of Pol η function. All of these trends in binding free energies are consistent with experimental measurements of binding strength, fidelity, processivity, and overall efficiency. The insights gained from this analysis have implications for drug design efforts aimed at modifying the binding properties of this enzyme for improving cancer chemotherapy treatments.
Comparative Molecular Dynamics Studies of Human DNA Polymerase η
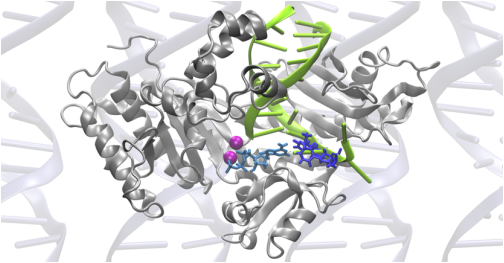
High-energy ultraviolet radiation damages DNA through the formation of cyclobutane pyrimidine dimers, which stall replication. When the lesion is a thymine-thymine dimer (TTD), human DNA polymerase η (Pol η) assists in resuming the replication process by inserting nucleotides opposite the damaged site. We performed extensive molecular dynamics (MD) simulations to investigate the structural and dynamical effects of four different Pol η complexes with or without a TTD and with either dATP or dGTP as the incoming base. No major differences in the overall structures and equilibrium dynamics were detected among the four systems, suggesting that the specificity of this enzyme is due predominantly to differences in local interactions in the binding regions. Analysis of the hydrogen-bonding interactions between the enzyme and the DNA and dNTP provided molecular-level insights. Specifically, the TTD was observed to engage in more hydrogen-bonding interactions with the enzyme than its undamaged counterpart of two normal thymines. The resulting greater rigidity and specific orientation of the TTD are consistent with the experimental observation of higher processivity and overall efficiency at TTD sites than at analogous sites with two normal thymines. The similarities between the systems containing dATP and dGTP are consistent with the experimental observation of relatively low fidelity with respect to the incoming base. Moreover, Q38 and R61, two strictly conserved amino acids across the Pol η family, were found to exhibit persistent hydrogen-bonding interactions with the TTD and cation-p interactions with the free base, respectively. Thus, these simulations provide molecular level insights into the basis for the selectivity and efficiency of this enzyme, as well as the roles of the two most strictly conserved residues.
High-energy ultraviolet radiation damages DNA through the formation of cyclobutane pyrimidine dimers, which stall replication. When the lesion is a thymine-thymine dimer (TTD), human DNA polymerase η (Pol η) assists in resuming the replication process by inserting nucleotides opposite the damaged site. We performed extensive molecular dynamics (MD) simulations to investigate the structural and dynamical effects of four different Pol η complexes with or without a TTD and with either dATP or dGTP as the incoming base. No major differences in the overall structures and equilibrium dynamics were detected among the four systems, suggesting that the specificity of this enzyme is due predominantly to differences in local interactions in the binding regions. Analysis of the hydrogen-bonding interactions between the enzyme and the DNA and dNTP provided molecular-level insights. Specifically, the TTD was observed to engage in more hydrogen-bonding interactions with the enzyme than its undamaged counterpart of two normal thymines. The resulting greater rigidity and specific orientation of the TTD are consistent with the experimental observation of higher processivity and overall efficiency at TTD sites than at analogous sites with two normal thymines. The similarities between the systems containing dATP and dGTP are consistent with the experimental observation of relatively low fidelity with respect to the incoming base. Moreover, Q38 and R61, two strictly conserved amino acids across the Pol η family, were found to exhibit persistent hydrogen-bonding interactions with the TTD and cation-p interactions with the free base, respectively. Thus, these simulations provide molecular level insights into the basis for the selectivity and efficiency of this enzyme, as well as the roles of the two most strictly conserved residues.
Structure and Dynamics of Intrinsically Disordered Proteins
Models for the Metal Transfer Complex of the N-terminal Region of CusB and CusF
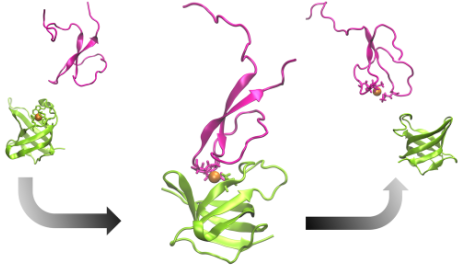
The tripartite CusCFBA pump in Escherichia coli is a very effective heavy metal extrusion system specific for Cu(I) and Ag(I). The N-terminal region of the membrane fusion protein CusB (CusB-NT) is highly disordered and, hence, experimentally characterizing its structure is challenging. In a previous study, this disorder was confirmed with molecular dynamics simulations although some key structural elements were determined. It was experimentally shown that CusB-NT is fully functional in transferring the metal from the metallochaperone CusF. In this study, we docked these two entities together and formed two representative metal coordination modes, which consist of residues from both proteins. In this way we created two potential CusB-NT/CusF complexes which share coordination of Cu(I) and thereby represent structural models for the metal transfer process. Each model complex was simulated for 4 µs. The previously observed structural disorder in CusB-NT disappeared upon complexation with CusF. The only differences between the two models occurred in the M21-M36 loop region of CusB-NT and the open flap of CusF: we observed the model with two CusB-NT methionine residues and a CusF methionine as the metal coordination site (termed “MMM”) to be more stable than the model with a CusB-NT methionine, a CusF methionine, and a CusF histidine ligating the metal (termed “MMH”). The observed stability of the MMM model was probed for an additional 2 µs, yielding a total simulation time of 6 µs. We hypothesize that both MMM and MMH configurations might take part in the metal exchange process where the MMH configuration would show up first and would be followed by the MMM configuration. Given the experimental finding of comparable binding affinities of CusB-NT and CusF, the increased stability of the MMM configuration might be a determinant for the transfer from CusF to CusB-NT. The metal would be transferred from the more CusF-dominated metal binding environment (MMH model) to a more CusB-dominated one (MMM model) where the coordination environment is more stable. From the MMM model, the metal ion would ultimately be coordinated by the CusB methionines only, which would complete the Cu(I) transfer process.
The tripartite CusCFBA pump in Escherichia coli is a very effective heavy metal extrusion system specific for Cu(I) and Ag(I). The N-terminal region of the membrane fusion protein CusB (CusB-NT) is highly disordered and, hence, experimentally characterizing its structure is challenging. In a previous study, this disorder was confirmed with molecular dynamics simulations although some key structural elements were determined. It was experimentally shown that CusB-NT is fully functional in transferring the metal from the metallochaperone CusF. In this study, we docked these two entities together and formed two representative metal coordination modes, which consist of residues from both proteins. In this way we created two potential CusB-NT/CusF complexes which share coordination of Cu(I) and thereby represent structural models for the metal transfer process. Each model complex was simulated for 4 µs. The previously observed structural disorder in CusB-NT disappeared upon complexation with CusF. The only differences between the two models occurred in the M21-M36 loop region of CusB-NT and the open flap of CusF: we observed the model with two CusB-NT methionine residues and a CusF methionine as the metal coordination site (termed “MMM”) to be more stable than the model with a CusB-NT methionine, a CusF methionine, and a CusF histidine ligating the metal (termed “MMH”). The observed stability of the MMM model was probed for an additional 2 µs, yielding a total simulation time of 6 µs. We hypothesize that both MMM and MMH configurations might take part in the metal exchange process where the MMH configuration would show up first and would be followed by the MMM configuration. Given the experimental finding of comparable binding affinities of CusB-NT and CusF, the increased stability of the MMM configuration might be a determinant for the transfer from CusF to CusB-NT. The metal would be transferred from the more CusF-dominated metal binding environment (MMH model) to a more CusB-dominated one (MMM model) where the coordination environment is more stable. From the MMM model, the metal ion would ultimately be coordinated by the CusB methionines only, which would complete the Cu(I) transfer process.
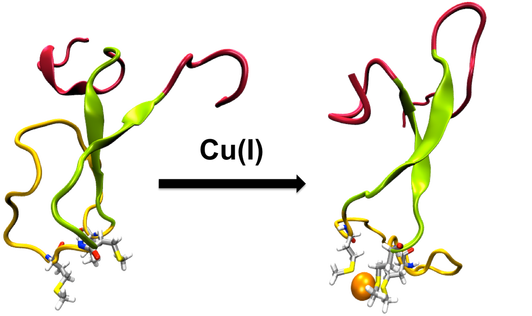
The N-terminal Region of the Cu(I)-binding Protein CusB
CusCFBA is one of the metal efflux systems in Escherichia coli that is highly specific for its substrates, Cu(I) and Ag(I). It serves to protect the bacteria in environments that have lethal concentrations of these metals. The membrane fusion protein CusB is the periplasmic piece of CusCFBA, which has not been fully characterized by crystallography because of its extremely disordered N-terminal region. This region has both structural and functional importance because it has been experimentally proven to transfer the metal by itself from the metallochaperone CusF and to induce a structural change in the rest of CusB to increase Cu(I)/Ag(I) resistance. Understanding metal uptake from the periplasm is critical to gain insight into the mechanism of the whole CusCFBA pump, which makes resolving a structure for the N-terminal region necessary because it contains the metal binding site. We ran extensive molecular dynamics simulations to reveal the structural and dynamic properties of both the apo and Cu(I)-bound versions of the CusB N-terminal region. In contrast to its functional companion CusF, Cu(I) binding to the N-terminus of CusB causes only a slight, local stabilization around the metal site. The trajectories were analyzed in detail, revealing extensive structural disorder in both the apo and holo forms of the protein. CusB was further analyzed by breaking the protein up into three subdomains according to the extent of the observed disorder: the N- and C-terminal tails, the central beta strand motif, and the M21–M36 loop connecting the two metal-coordinating methionine residues. Most of the observed disorder was traced back to the tail regions, leading us to hypothesize that the latter two subdomains (residues 13–45) may form a functionally competent metal-binding domain because the tail regions appear to play no role in metal binding.
CusCFBA is one of the metal efflux systems in Escherichia coli that is highly specific for its substrates, Cu(I) and Ag(I). It serves to protect the bacteria in environments that have lethal concentrations of these metals. The membrane fusion protein CusB is the periplasmic piece of CusCFBA, which has not been fully characterized by crystallography because of its extremely disordered N-terminal region. This region has both structural and functional importance because it has been experimentally proven to transfer the metal by itself from the metallochaperone CusF and to induce a structural change in the rest of CusB to increase Cu(I)/Ag(I) resistance. Understanding metal uptake from the periplasm is critical to gain insight into the mechanism of the whole CusCFBA pump, which makes resolving a structure for the N-terminal region necessary because it contains the metal binding site. We ran extensive molecular dynamics simulations to reveal the structural and dynamic properties of both the apo and Cu(I)-bound versions of the CusB N-terminal region. In contrast to its functional companion CusF, Cu(I) binding to the N-terminus of CusB causes only a slight, local stabilization around the metal site. The trajectories were analyzed in detail, revealing extensive structural disorder in both the apo and holo forms of the protein. CusB was further analyzed by breaking the protein up into three subdomains according to the extent of the observed disorder: the N- and C-terminal tails, the central beta strand motif, and the M21–M36 loop connecting the two metal-coordinating methionine residues. Most of the observed disorder was traced back to the tail regions, leading us to hypothesize that the latter two subdomains (residues 13–45) may form a functionally competent metal-binding domain because the tail regions appear to play no role in metal binding.
Novel Free Energy estimation Methods with on-the-Fly Error Estimation
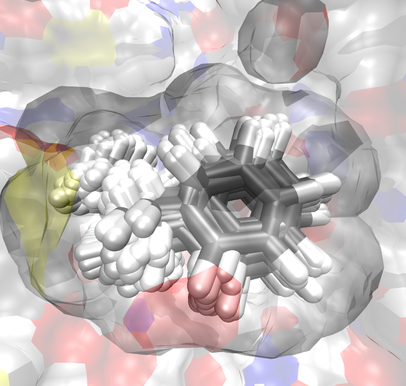
Bringing Clarity to the Prediction of Protein–Ligand Binding Free Energies via “Blurring”
We present a novel method to evaluate the free energies of ligand binding utilizing a Monte Carlo estimation of the configuration integrals concomitant with uncertainty quantification. Ensembles for integration are built through systematically perturbing an initial ligand conformation in a rigid binding pocket, which is optimized separately prior to incorporation of the ligand. The procedure producing the ensembles is called “blurring” and is carried out using in-house built code. The Boltzmann factor contribution of each pose to the configuration integral is computed and from there the free energy is obtained. Potential function uncertainties are estimated using a fragment-based error propagation method. This method has been applied to a set of small aromatic ligands complexed with T4 Lysozyme L99A mutant. Microstate scoring has been carried out with the ff99SB, ff94, and PM6DH2 functionals in conjunction with continuum solvation models like Generalized Born (GB), Conductor-like Screening Model (COSMO), and SMD. Especially PM6DH2 scoring has yielded a binding free energy estimate which is in good correlation to the experimental binding affinities (R2=0.7) whereas the calculated binding free energies have been underestimated with all methods. We suggest this is due to the single static protein structure and inaccuracies in the solvent models we have employed.
We present a novel method to evaluate the free energies of ligand binding utilizing a Monte Carlo estimation of the configuration integrals concomitant with uncertainty quantification. Ensembles for integration are built through systematically perturbing an initial ligand conformation in a rigid binding pocket, which is optimized separately prior to incorporation of the ligand. The procedure producing the ensembles is called “blurring” and is carried out using in-house built code. The Boltzmann factor contribution of each pose to the configuration integral is computed and from there the free energy is obtained. Potential function uncertainties are estimated using a fragment-based error propagation method. This method has been applied to a set of small aromatic ligands complexed with T4 Lysozyme L99A mutant. Microstate scoring has been carried out with the ff99SB, ff94, and PM6DH2 functionals in conjunction with continuum solvation models like Generalized Born (GB), Conductor-like Screening Model (COSMO), and SMD. Especially PM6DH2 scoring has yielded a binding free energy estimate which is in good correlation to the experimental binding affinities (R2=0.7) whereas the calculated binding free energies have been underestimated with all methods. We suggest this is due to the single static protein structure and inaccuracies in the solvent models we have employed.
Measuring the Effects of Non-additivity in Interaction Energy Calculations
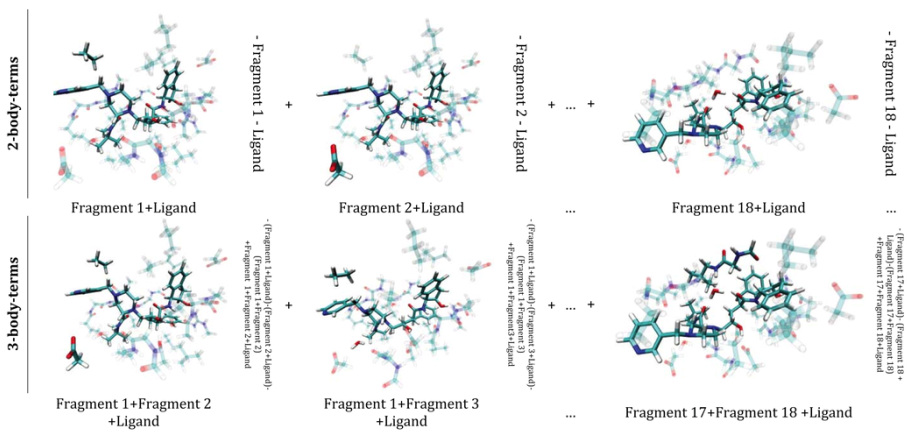
Pairwise additivity of energy components in protein-ligand binding: The HIV II protease-Indinavir case
An energy expansion (binding energy decomposition into n-body interaction terms for n ≥ 2) to express the receptor-ligand binding energy for the fragmented HIV II protease-Indinavir system is described to address the role of cooperativity in ligand binding. The outcome of this energy expansion is compared to the total receptor-ligand binding energy at the Hartree-Fock, density functional theory, and semi-empirical levels of theory. We find that the sum of the pairwise interaction energies approximates the total binding energy to ∼82% for HF and to >95% for both the M06-L density functional and PM6-DH2 semiempirical method. The contribution of the three-body interactions amounts to 18.7%, 3.8%, and 1.4% for HF, M06-L, and PM6-DH2, respectively. We find that the expansion can be safely truncated after n = 3. That is, the contribution of the interactions involving more than three parties to the total binding energy of Indinavir to the HIV II protease receptor is negligible. Overall, we find that the two-body terms represent a good approximation to the total binding energy of the system, which points to pairwise additivity in the present case. This basic principle of pairwise additivity is utilized in fragment-based drug design approaches and our results support its continued use. The present results can also aid in the validation of non-bonded terms contained within common force fields and in the correction of systematic errors in physics-based score functions.
